Blask w kolbie
Poniższy artykuł został opublikowany pierwotnie w czasopiśmie dla nauczycieli Chemia w Szkole (3/2023):

Myślę, że nie muszę przekonywać Czytelnika, że chemia jest niezwykle fascynującą dziedziną nauki, która bada skład, właściwości i przemiany otaczającej nas materii. Obejmuje szeroki zakres dyscyplin, od chemii organicznej i nieorganicznej po chemię fizyczną i analityczną, oferując ogromną przestrzeń do zdobywania wiedzy. Ze swoją fundamentalną rolą w naszym codziennym życiu, chemia daje unikalne spojrzenie na otaczający nas świat.
Jednym z fascynujących aspektów chemii są procesy chemiluminescencyjne, w których światło jest emitowane w wyniku reakcji chemicznej. Powstające w ten sposób światło może mieć różny kolor, intensywność i trwałość, tworząc wizualnie zachwycające widowiska, które fascynują zarówno uczniów, jak i mogą być przydatne w bardziej naukowych celach.
Wykorzystanie reakcji chemiluminescencyjnych ma duży potencjał w dydaktyce. Poprzez wprowadzanie tych reakcji do zajęć edukacyjnych, nauczyciele mogą angażować uczniów w sposób ciekawy i interaktywny. Efektowne emisje świetlne mogą wzbudzać ciekawość, a przez to ułatwiać głębsze zrozumienie procesów chemicznych. Warto zauważyć, że dzięki widowiskowemu efektowi tego typu reakcje sprawiają, że nawet stosunkowo skomplikowane i abstrakcyjne koncepcje - jak choćby przemiany energii - stają się bardziej dostępne i zapadające w pamięć. Co więcej, z punktu widzenia dydaktyki tematyka reakcji chemiluminescencyjnych jest bardzo wszechstronna: od demonstracji kinetyki reakcji i transferu energii, po badanie wpływu różnych reagentów i warunków na przebieg reakcji.
Istnieje wiele substancji o właściwościach chemiluminescencyjnych – jedną z najbardziej znanych jest luminol C8H7N3O2 i lofina C21H16N2 [1] [2]. Dziś jednak chciałbym jednak opisać moje prace w kierunku syntezy i obserwacji chemiluminescencji lucygeniny, a więc dużo mniej znanego związku o podobnych właściwościach. Zachęcam oczywiście Czytelnika do własnych prac w tym zakresie i prób powtórzenia opisanych tutaj reakcji.
Opisana tutaj metoda syntezy bazuje na protokole opublikowanym w 1982 roku, z pewnymi modyfikacjami [3].
Nie przedłużając, przejdźmy więc do pierwszego etapu syntezy.
Etap I – Kwas N-fenyloantranilowy
Rozpoczynając nasze prace musimy zgromadzić substancje takie jak:
- kwas 2-chlorobenzoesowy (o-chlorobenzoesowy) ClC6H4CO2H,
- anilina C6H7N,
- węglan potasu bezwodny K2CO3,
- tlenek miedzi(I) Cu2O,
- węgiel aktywny C,
- kwas chlorowodorowy HCl(aq).
Kwas 2-chlorobenzoesowy jest jednym z trzech izomerów kwasu chlorobenzoesowego i posiada wśród nich najsilniejsze właściwości kwasowe. W warunkach normalnych jest białym ciałem krystalicznym i służy jako substrat do syntezy wielu leków, dodatków do żywności i barwników [4]. Związek ten działa drażniąco na skórę, oczy i drogi oddechowe.
Z kolei anilina jest najprostszą aminą aromatyczną. Występuje ona jako bezbarwna ciecz, brunatniejąca powoli na powietrzu, o charakterystycznym zapachu mogącym kojarzyć z zepsutą rybą. Jej gęstość jest wyraźnie większa od gęstości wody, w której jest słabo rozpuszczalna. Zastosowanie aniliny jest bardzo szerokie w przemyśle chemicznym, farmaceutycznym, barwnikarskim i gumowym, a także przy produkcji materiałów wybuchowych i jako składnik niektórych paliw rakietowych.
Chcę wyraźnie zaznaczyć, że anilina jest substancją trującą. Działa toksycznie na skutek każdego rodzaju narażenia: m.in. przez drogi oddechowe, po połknięciu i w kontakcie ze skórą. W następstwie długotrwałego kontaktu z nawet niewielkimi ilościami tego związku powstaje poważne zagrożenie dla zdrowia. Anilina szczególnie drastycznie działa na krew i układ krwiotwórczy - między innymi niszczy czerwone krwinki. Trzeba brać także pod uwagę jej prawdopodobną aktywność mutageniczną.
Tlenek miedzi(I) w przyrodzie występuje jako czerwony minerał kupryt i przez długi czas był jednym z głównych źródeł miedzi dla człowieka.
Węglan potasu w warunkach normalnych ma postać białego ciała krystalicznego, dobrze rozpuszczalnego w wodzie. Posiada stosunkowo wysoką temperaturę topnienia (według różnych źródeł 891–899°C). Znany jest od starożytności jako potaż, uzyskiwany z popiołu drzewnego poprzez ługowanie.
Przystępując do syntezy, w kolbie okrągłodennej umieściłem 20g kwasu o-chlorobenzoesowego, 80g świeżo przedestylowanej aniliny, 20g bezwodnego węglanu potasu i 0,5g tlenku miedzi(I). Kolba wraz z chłodnicą zwrotną została umieszczona na czaszy grzejnej, po czym brunatną mieszaninę stanowiącą jej zawartość ogrzewałem do wrzenia przez 2,5 godziny (Fot.1). Po tym czasie ogrzewanie zostało wyłączone, a mieszanina poreakcyjna ochłodzona do temperatury pokojowej i następnie zadana 300cm3 wody.

Na tym etapie musimy się pozbyć z mieszaniny poreakcyjnej pozostałości nieprzereagowanej aniliny. Można to zrobić na różne sposoby, między innymi przez odpędzenie poprzez destylację z parą wodną. Proces ten może być jednak dla wielu eksperymentatorów kłopotliwy, więc można go zastąpić przez kilkukrotną ekstrakcję wspomnianej aminy niewielkimi porcjami eteru dietylowego C4H10O (uwaga, łatwopalny!) w rozdzielaczu. Zadanie to jest niestety dosyć niewdzięczne, ponieważ zarówno spodnia faza wodna (zawierająca produkt), jak i górna faza eterowa są ciemnobrunatne (prawie czarne). Z tego powodu zauważenie granicy między nimi jest dosyć trudne. Przydatne jest tu silne oswietlenie boczne, które ułatwia rozpoznanie tej granicy (Fot.2).

Pozbawiony już w dużej mierze śladów aniliny roztwór wodny podgrzewa się do wrzenia z dodatkiem 10 g węgla aktywnego C w czasie kilkunastu minut, po czym sączy na gorąco. Przesącz po ochłodzeniu łączy się z 60 cm3 kwasu chlorowodorowego o stężeniu kilkunastu procent (u mnie 14%), co powoduje wydzielenie się dużych ilości osadu (Fot.3). Jest to kwas N-fenyloantranilowy powstały w reakcji nukleofilowego podstawienia kwasu o-chlorobenzoesowego aniliną (Rys.1).

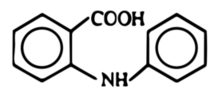
Produkt należy odsączyć i wysuszyć w eksykatorze. Kwas N-fenyloantranilowy w postaci czystej ma postać białych kryształów, ale zsyntezowany opisaną metodą posiada często delikatnie szare zabarwienie (Fot.4). Jego czystość jest wystarczająca do przeprowadzenia dalszych etapów syntezy.

Uzyskano około 23g wspomnianego kwasu przy opisanej proporcji substratów, co stanowi ~84% procent wydajności teoretycznej w stosunku do użytego kwasu 2-chlorobenzoesowego. Do dalszych syntez można wykorzystać całą ilość kwasu, lub pewną jego część pozostawić w innym celu.
Etap II – Od kwasu N-fenyloantranilowego do akrydonu
Ten krok syntezy wymaga jedynie substancji takich jak:
- kwas N-fenyloantranilowy C13H11NO2,
- kwas siarkowy(VI) H2SO4 stężony,
- węglan sodu Na2CO3.
Kwas N-fenyloantranilowy znajduje dosyć szerokie zastosowanie jako pólprodukt w otrzymywaniu farmaceutyków, a także w syntezie peptydów. Związek ten ma właściwości drażniące. Kwas siarkowy(VI) z kolei ma silne właściwości żrące i przy bezpośredniej ekspozycji z łatwością niszczy tkanki naszego ciała.
Chcąc przeprowadzić kwas N-fenyloantranilowy w kolejny półprodukt rozpuściłem 20g tej substancji w 44cm3 stężonego kwasu siarkowego(VI) i ogrzewałem na łaźni parowej przez 1,5 godziny, otrzymując ciemnozielony roztwór. Następny etap jest dosyć niebezpieczny i wymaga maksymalnej uwagi przy jego przeprowadzaniu. Jeszcze gorący, zielony roztwór należy wlać porcjami i bardzo ostrożnie do około 150 cm3 wody o temperaturze bliskiej wrzenia, co może powodować liczne rozpryski silnie żrącej mieszaniny. Dlatego reakcję najlepiej jest prowadzić na dnie wysokiej zlewki o pojemności 1l lub większej. Konieczne jest także zastosowanie ochrony oczu i całej twarzy, np. w postaci maski z tworzywa sztucznego. Po dodaniu całej mieszaniny reakcyjnej do wody powstaje zawiesina o brudnożółtawej barwie, którą należy ogrzewać do wrzenia w czasie 5 minut i przesączyć. Osad dodaje się bez suszenia do 200cm3 roztworu węglanu sodu (należy wziąć pod uwagę, że mieszanina silnie się pieni z racji wywiązywanego gazu) o stężeniu około 8% i ogrzewa przez kilka minut ponownie do wrzenia.
Zbyt duża ilość węglanu może spowodować roztworzenie osadu, w takim przypadku warto dodawać roztwór zasady porcjami do uzyskania odczynu zbliżonego do neutralnego względem papierka uniwersalnego (przyp. aut.).
Po odsączeniu uzyskujemy bezpostaciowy surowy akrydon (Fot.5) powstały na drodze wewnątrzcząsteczkowego podstawienia elektrofilowego pod wpływem stężonego kwasu siarkowego(VI).

Niestety praktyka pokazuje, że uzyskany w ten sposób akrydon nie może być wykorzystany w dalszych etapach syntezy bez znaczącego spadku wydajności. Aby tego uniknąć, musimy oczyścić substancję przez rekrystalizację. Problemem jest jednak fakt, że akrydon bardzo słabo rozpuszcza się w łatwodostępnych rozpuszczalnikach i klasyczna rekrystalizacja byłaby w tym przypadku mało wydajna. Rozwiązaniem jest zastosowanie aparatu Soxhleta, który jest przyrządem laboratoryjnym służącym do ekstrakcji trudno rozpuszczalnych związków chemicznych, wynalezionym w 1879 roku przez Franza von Soxhleta [5].
Aparat Soxhleta składa się z układu szklanych rurek i komory, które są włączone między kolbę z wrzącym rozpuszczalnikiem a chłodnicę zwrotną (Rys.3). Wrzące pary rozpuszczalnika a przemieszczają się z dolnej kolby przez rurkę b do chłodnicy c (z ciągłym obiegiem wody chłodzącej), która znajduje się powyżej aparatu. Próbka do ekstrakcji jest umieszczana w komorze ekstrakcyjnej d w specjalnym koszyku, czyli tzw. gilzie e wykonanej np. z celulozy. Na dolnym końcu komory, poniżej poziomu jej dna, na którym opiera się gilza, znajduje się wylot syfonu f. Gdy poziom cieczy w komorze ekstrakcyjnej napełnianej skroplonym rozpuszczalnikiem osiąga pewien poziom okreslony przez budowę syfonu, cała zgromadzona ciecz samoczynnie wypływa przez tę rurkę do kolby.
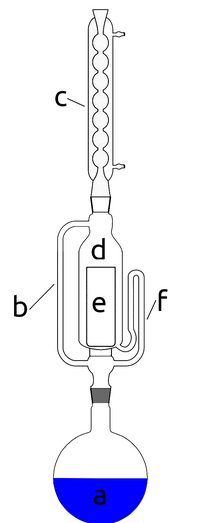
Warto zauważyć, że cały układ pracuje więc cyklicznie, a komora ekstrakcyjna powoli wypełnia się świeżo przedestylowanym rozpuszczalnikiem do górnego poziomu syfonu, a gdy ten poziom zostaje osiągnięty, aparat opróżnia się automatycznie i ponownie napełnia świeżym rozpuszczalnikiem.
Zauważmy, że nawet jeśli w jednym cyklu rozpuszczeniu ulegnie jedynie bardzo niewielka ilość substancji znajdującej się w gilzie, to dzięki cyklicznej pracy zostanie ona powoli w całości przeniesiona do dolnej kolby, pozostawiając nierozpuszczalne zanieczyszczenia w komorze ekstrakcyjnej. Pozwala to niejako wykorzystać wielokrotnie tą samą objętość rozpuszczalnika, a tym samym ograniczyć jego ilość.
Przystępując do pracy umieściłem surowy akrydon w gilzie o odpowiednio dobranym rozmiarze (Fot.6).

Następnie zestawiłem aparat Soxhleta i umieściłem w nim gilzę, ustawiając ją na przekładce z niewielkiej ilości waty. Bardzo istotną sprawą jest, aby górna krawędź gilzy była umieszczona powyżej zagięcia syfonu (Fot.7).

Jako rozpuszczalnik korzystnie jest zastosować w tym przypadku etanol o stężeniu co najmniej 95%. Jego ilość trzeba dobrać doświadczalnie – w moim przypadku było to około 400cm3.
Aparat Soxhleta wyraźnie przyspiesza i ułatwia oczyszczenie akrydonu, jest to jednak w dalszym ciągu proces stosunkowo powolny. Przy podanych ilościach substancji w moim przypadku trwało to kilkanaście godzin, podczas których mieszanina w kolbie przez cały czas wrzała.
Rozpuszczalność akrydonu w alkoholu jest tak nikła, że już po kilku cyklach pracy aparatu można było zauważyć krystalizację z roztworu w kolbie ze wrzącym rozpuszczalnikiem.
Proces przerwałem po stwierdzeniu, że w gilzie pozostały już tylko nierozpuszczalne pozostałości. Po ochłodzeniu mieszaninę z kolby przesączyłem, a zebrany osad wysuszyłem uzyskując piękne żółte kryształki oczyszczonego akrydonu (Fot.8).

Z podanej ilości kwasu N-fenyloantranilowego uzyskano około 16,3g surowego akrydonu, z czego po oczyszczeniu pozostało około 14,8g, co przekłada się odpowiednio na wydajności 89% i 81% w stosunku do wskazanego substratu.
Tak przygotowany akrydon może zostać wykorzystany w kolejnym etapie.
Etap III – Od akrydonu do N-metyloakrydonu
Na tym etapie potrzebujemy nieco więcej substancji. Są to:
- akrydon C13H9NO,
- wodorotlenek potasu KOH,
- etanol C2H5OH,
- dimetyloformamid C3H7NO,
- jodometan (jodek metylu) CH3I.
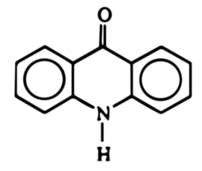
Akrydon jest związkiem organicznym o strukturze opartej na szkielecie akrydyny (Rys. 2). Jak już widzieliśmy, substancja ta w warunkach normalnych ma postać żółtego ciała krystalicznego. Niektóre pochodne akrydonu są używane jako znaczniki fluorescencyjne w biologii molekularnej. Trzeba wspomnieć, że substancja ta jest podstawowym substratem do syntezy wielu związków stosowanych w lecznictwie. Akrydon ma działanie drażniące.
Dimetyloformamid jest organicznym związkiem chemicznym z grupy amidów. W warunkach normalnych jest to ciecz mieszająca się w dowolnym stosunku z wodą oraz wieloma rozpuszczalnikami organicznymi. Czysta substancja jest pozbawiona zapachu, ale obecność dimetyloaminy C2H7N w produkcie klasy technicznej nadaje mu nieprzyjemny, rybny zapach. Jest powszechnie stosowany jako rozpuszczalnik o przydatnych właściwościach; cząsteczka dimetylormamidu ma charakter polarny i hydrofilowy. Dzięki temu prowadzenie reakcji w środowisku dimetyloformamidu ułatwia zachodzenie wielu reakcji, np. substytucji nukleofilowej [6]. Należy zachować szczególną ostrożność przy pracy z tą substancją, ponieważ podejrzewa się ją o własności karcynogenne i teratogenne.
Kolejna substancja, jaką jest jodometan lub inaczej jodek metylu, to organiczny związek chemiczny z grupy halogenków alkilowych, pojedyńczo podstawiona jodem pochodna metanu. Stanowi bezbarwną ciecz, powoli brunatniejąca pod wpływem światła z powodu rozkładu, czego jednym z produktów jest nadający wspomniane zabarwienie jod. Temperatura topnienia związku to −66°C, a temperatura wrzenia 42,4°C. Substancja ta jest słabo rozpuszczalna w wodzie, rozpuszczalna z kolei w alkoholu etylowym i eterze dietylowym. Jodometan stosuje się głównie w syntezie organicznej i przemyśle farmaceutycznym do metylowania, którą to możliwość wykorzystamy w syntezie także i my. Związek ten jest silnie toksyczny i lotny. Wszelkie manipulacje z nim muszą być wykonywane pod sprawnie działającym wyciągiem.
Przystępując to aktu syntezy rozpuściłem na gorąco 10g akrydonu w 122 cm3 etanolu z dodatkiem 3,15g wodorotlenku potasu. Etanol następnie trzeba odparować, co najlepiej zrobić w rotacyjnej wyparce próżniowej, ale przetestowałem także sposób nie wymagający posiadania tego urządzenia. Jak stwierdziłem, całkowicie wystarczające jest odparowanie etanolu w parowniczce lub krystalizatorze na łaźni parowej, dbając jednak aby nie zawilgocić mieszaniny zawierającej higroskopijny wodorotlenek. W ten sposób uzyskano żółtą pozostałość (Fot.9), którą następnie rozpuściłem w 122cm3 dimetyloformamidu uzyskując ciemnozielony roztwór (Fot.10).


Do roztworu dodałem następnie po kropli 8,52g jodometanu, po czym ogrzałem na łaźni parowej w czasie 15 minut, co spowodowało niewielką zmianę – płyn przestał barwić ścianki naczynia (Fot.11).

W opisanym procesie dochodzi do nukleofilowego podstawienia jodku metylu anionem akrydynowym. Po wlaniu mieszaniny rakcyjnej do wody uzyskuje się jasnożółty osad surowego N-metyloakrydonu (Fot.12).

Osad po odsączeniu i wysuszeniu wydaje się być mało efektowną, kremowożółtą bezpostaciową substancją (Fot.13).

Substancję oczyściłem następnie przez klasyczną rekrystalizację z gorącego etanolu. Podczas ochładzania stężonego roztworu pochodnej wydziela się ona w postaci igiełkowatych kryształów (Fot.14).

Po odsączeniu i wysuszeniu pochodna jest gotowa do dalszej obróbki (Fot.15).

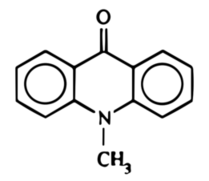
W czasie syntezy uzyskałem 9,32g surowego N-metyloakrydonu i 8,46 oczyszczonego przez rekrystalizację, co przełożyło się na wydajności odpowiednio 87% i 79% w stosunku do wykorzystanego akrydonu.
Etap III – Od N-metyloakrydonu do lucygeniny
W tym kroku potrzebujemy:
- N-metyloakrydon C14H11NO,
- etanol C2H5OH (~95%),
- kwas chlorowodorowy HCl(aq) stężony,
- cynk Zn,
- kwas azotowy(V) HNO3 ~6%.
N-metyloakrydon jest pochodną akrydonu uzyskaną przez jego metylację w opisanych warunkach i ma zastosowanie jako półprodukt w syntezach organicznych.
Wykorzystany przez nas cynk musi być w postaci jak najdrobniejszego proszku. Nadmienię, że silnie sproszkowany cynk ma dosyć silne właściwości brudzące, a także może być piroforyczny, czego nie należy zaniedbywać.
Przy manipulacjach z kwasami należy uważać jak zawsze przy pracy z tego rodzaju chemikaliami.
W kolbie okrągłodennej umieściłem 9g N-metyloakrydonu, 450cm3 etanolu i 90cm3 stężonego kwasu chlorowodorowego. Pełne rozpuszczenie substancji stałych wymaga ogrzewania pod chłodnicą zwrotną (uwaga na drażniące pary). Następnie do mieszaniny dodałem powoli 28.8g pyłu cynkowego, co zajęło około 40 minut. Mieszaninę ogrzewałem potem do wrzenia pod chłodnicą zwrotną w czasie kolejnej godziny co doprowadziło do uzyskania ciemnego roztworu (Fot.16).

Mieszaninę poreakcyjną po ochłodzeniu wlałem powolnym strumieniem i ciągle mieszając do 1l zimnej wody, a wydzielony zielony osad związku bisakrydynowego odsączyłem i wysuszyłem (Fot.17, Rys.5).

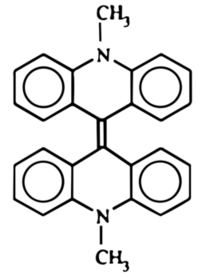
Zielony osad roztworzyłem potem w 540cm3 kwasu azotowego(V) o stężeniu około 6% i ogrzewałem na łaźni parowej przez 30 minut, po czym przesączyłen ciemny roztwór na gorąco i pozostawiłem do krystalizacji w temperaturze pokojowej na noc. Następnego dnia można było podziwiać wydzielone z roztworu pomarańczowoczerwone kryształy lucygeniny C28H22N4O6 (Fot.18).

Kryształy lucygeniny należy odsączyć i wysuszyć w niezbyt wysokiej temperaturze. Na sucho ich barwa jest nieco bardziej pomarańczowa niż czerwona (Fot.19).

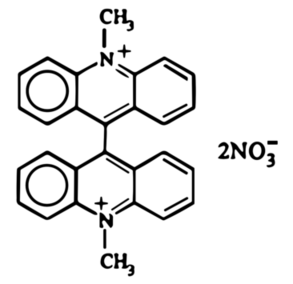
W ten sposób została przeprowadzona redukcyjna dimeryzacja N-metyloakrydonu do pochodnej bis-akrydynowej pod wpływem cynku w kwaśnym środowisku. Związek ten następnie uległ utlenieniu kwasem azotowym z utworzeniem odpowiedniego diazotanu. Z chemicznego punktu widzenia lucygenina ma więc postać rozpuszczalnego diazotanu bis-N-metyloakrydyny.
Uzyskałem 7,7g lucygeniny, co stanowi około 70% maksymalnej wydajności teoretycznej w przeliczeniu na wykorzystany N-metyloakrydon. Gotową lucygeninę należy przechowywać w naczyniu z ciemnego szkła.
Fluorescencja otrzymanych substancji
Część z otrzymanych na poszczegolnych etapach substancji wykazuje ciekawą cechę, jaką jest fluorescencja, czyli emisja światła z zakresu widzialnego pod wpływem wzbudzenia promieniowaniem ultrafioletowym. Może to być dodatkowym sprawdzianem, czy na każdym etapie uzyskaliśmy odpowiedni związek. W tym celu do kolejnych probówek należy wprowadzić po kilka mililitrów wody destylowanej i maleńkim kryształku każdej z substancji. Z racji niewielkiej rozpuszczalności niektórych z nich w każdym przypadku zawartość probówki wytrząsano przez 1 minutę, po czym zaobserwowano efekt po oświetleniu promieniowaniem UV (Fot.20). W ten sposób możemy się przekonać, że roztwór kwasu N-fenyloantanilowego w tych warunkach nie fluoryzuje lub emisja jest niezauważalna, akrydon i N-metyloakrydon fluoryzuje niebiesko (ten drugi nieco słabiej, ale w tak prostym doświadczeniu trudno wskazać jaki czynnik miał na to wpływ), natomiast przejściowa pochodna bis-akrydynowa i lucygenina fluoryzują bardzo silnie emitując światło o pięknej zielonej barwie.

Chemiluminescencja lucygeniny
Po trudach syntezy i powodzeniu na wszystkich etapach wreszcie nadchodzi ten moment, kiedy możemy wypróbować zdolność lucygeniny do generowania światła widzialnego podczas jej utleniania. W tym celu musimy przygotować dwa roztwory:
| A: | w 50cm3 wody rozpuścić małą szczyptę (0,05g) lucygeniny, |
| B: | w 35cm3 wody rozpuścić 15cm3 etanolu, 4g wodorotlenku sodu i 2,5cm3 nadtlenku wodoru o stężeniu 3% (aptecznej wody utlenionej). |
Roztwór A jest pomarańczowo-żółty, natomiast B całkowicie bezbarwny (Fot.21). Oba roztwory najlepiej jest przygotowywać na świeżo, można je jednak przez dosyć krótki czas przechowywać w lodówce.

Aby zaobserwować chemiluminescencję, warto zaciemnić jak najdokładniej pomieszczenie, po czym wlać szybkim ruchem całą objętość roztworu B do naczynia zawierającego roztwór A. Praktycznie natychmiast rozpoczyna się mogąca trwać kilkanaście minut emisja zielonego światła (Fot.22).

Co ciekawe, przy dluższej obserwacji można zauważyć, że podczas postępującego zużycia substratów poza słabnięciem z czasem emisji zmienia się także barwa powstającego światła – zyskuje ono coraz wyraźniejszy odcień niebieski (Fot.23).

Wyjaśnienie
Lucygenina ma silne właściwości chemiluminescencyjne podczas reakcji jej utleniania nadtlenkiem wodoru w środowisku wodnym o odczynie zasadowym. Najbardziej prawdopodobny mechanizm reakcji obejmuje utlenienie lucygeniny do nietrwałego cyklicznego nadtlenku, który nastepnie ulega rozpadowi do N-metyloakrydonu. Ten ostatni występuje jednak początkowo w metastabilnym stanie wzbudzonym, przez co ulega spontanicznemu przekształceniu do stanu podstawowego oddając nadmiarową energię. W początkowych fazach reakcji, kiedy w dużych ilościach dostępna jest jeszcze nieprzereagowana lucygenina, zostaje na nią przekazana energia wzbudzenia z cząsteczek N-metyloakrydonu - dlatego obserwujemy wtedy emisję zielonego światła charakterystycznego dla relaksujących cząsteczek tego związku (vide Fot.20 e). Wraz ze spadkiem dostępności lucygeniny, a więc i opisanego transferu energii, w widmie emisji zaczyna przeważać światło niebieskie, charakterystyczne dla wzbudzonego akrydonu i N-metyloakrydonu (Fot.20 b, c). W ten sposób opisane doświadczenie pozwala się w naoczny sposób przekonać o prawidłowościach rządzących przemianami materii i energii na poziomie molekularnym.
Zastosowania lucygeniny nie ograniczają się jedynie do dydaktyki. Zarówno ta substancja, jak i jej pochodne są wykorzystywane np. jako znaczniki molekularne w badaniach biologicznych.
Literatura:
- [1] Ples M., Widmowy blask. Chemiluminescencja katalizowana kompleksem miedzi, Chemia w Szkole, 2 (2016), Agencja AS Józef Szewczyk, str. 13-17 powrót
- [2] Ples M., Synteza i chemiluminescencja lofiny - zimne światło, muzyka i migdały, Chemia w Szkole, 5 (2020), Agencja AS Józef Szewczyk, str. 44-47 powrót
- [3] Amiet R.G., The preparation of lucigenin: An experiment with charm, Journal of Chemical Education, 59(2), 1982, str. 163-164 powrót
- [4] Maki T., Takeda K., Benzoic Acid and Derivatives, in: Ullmann's Encyclopedia of Industrial Chemistry, 2002, Wiley, Weinheim powrót
- [5] Harwood L. M., Moody Ch.J., Experimental organic chemistry: Principles and Practice, Wiley-Blackwell, 1989, str. 122-125 powrót
- [6] Mohammadkhani L., Beyond a solvent: triple roles of dimethylformamide in organic chemistry, RSC Advances, 8 (49), 2018, str. 27832-27862 powrót
Wszystkie fotografie i rysunki zostały wykonane przez autora
Marek Ples